The Evidence for Common Descent from Molecular Genetics - What John Watts' Article Never Mentioned
In order to see how poor John Watts' attempted rebuttal [1] of the evidence for common descent from molecular genetics was, it's helpful to summarise this evidence, if only to see how embarrassingly bad Watts' response was. The evidence for common descent from molecular genetics falls into two main groups: (1) sequence homology and (2) high degree of synteny conservation between related species. Again, while the precise details are quite complex, the underlying principles are easy to grasp.
Synteny

Shared genomic errors


1. Watts J "Evolution or Creation? The Argument From Comparative Anatomy" The Testimony January 2005 p 31-33
Two gene or protein sequences that are believed to share a common ancestor (either via speciation or gene duplication) are regarded as homologous. [2] To understand why gene similarity is viewed in terms of common descent rather than common design, we need to see how proteins are encoded in DNA.
Proteins are polymers of simpler molecules known as amino acids. The universal genetic code [3] defines how genetic material in the DNA is translated into proteins. It does this by defining how sequences of three nucleotides [4] called codons specify each of the 20 amino acids encoded by the genetic code.
When the cell needs to synthesise a protein, an RNA copy of the DNA code for that required protein is transcribed in a process known as transcription. The RNA transcript then moves to a large structure known as a ribosome. Here, small sections of RNA called transfer RNA (which carry on one end the amino acid coded by the three nucleotide sequence on the other end) pair to the matching codon on the mRNA.
As there are 4^3 or 64 possible codons, there is more than one way to code for each amino acid, making the genetic code redundant. On average, there are around 3 ways to encode for each amino acid.
The significance of this for common descent should be clear when we recognise just how vast the possible space for encoding the same gene exists due to the redundancy of the genetic code. Take the protein cytochrome c for example, which consists of around 104 amino acids, and is crucial in cellular respiration. There are 3^104 or 4.1 x 10^49 possible ways to encode for the same 104 amino acid sequence. As each of these possible genes encodes for the same protein, there is no a priori reason for a designer to use one of these possible genes over another. Conversely, common descent would predict that the cytochrome c gene sequences would reflect a pattern of descent with modification, with species sharing a remote common ancestor differing from each other by more neutral mutations (mutations in the codon that due to the redundancy of the genetic code do not change the amino acid for which it encodes) than those that share a more recent common ancestor.
This is in fact the pattern we see when we examine the cytochrome c sequences for multicellular life from yeast to human. Given the incredibly large coding space available in a protein such as cytochrome c, a designer could easily give every living creature its own unique gene sequence separated from all other species by a considerable amount of genetic space. This would neatly refute common descent – the fact that an evolutionary family tree plotted from cytochrome c data is consistent with the established evolutionary family tree derived from morphological data is, as the geneticist Theodore Dobzhansky pointed out, impossible to understand except in the light of evolution. [5]
Special creationist attempts to evade the force of this argument are unconvincing. We know that the cytochrome c molecules for humans will work when inserted into yeast cells – this is even more powerful when we recognise that the amino acids in cytochrome c can vary considerably without it losing functionality. The net result of both protein and coding redundancy is to extend the possible space of functional cytochrome c molecules to around 10^90. Despite this phenomenally large space, cytochrome c amino acid and gene sequences cluster in a pattern consistent with common descent.
Similar evolutionary trees can be constructed with other genes, and they are generally consistent with the standard evolutionary tree. Again, there is no reason one would expect this if life had been specially created – a designer could have specially created life with the nuclear genomic data clustering in a way to simulate common descent, but this raises the valid question as to why a creator would create within the genomic data of all life the appearance of an evolutionary origin.
Synteny
Another prediction of common descent is that closely related species should also share the same order of genes on their chromosomes. Geneticists use the term ‘synteny’ to describe the order of genes between the genomes of two species. A good analogy is that of two bookshelves with the same books on each shelf, arranged in the same order. Species with a high synteny have more genes in the same order. After speciation and divergence of the two descendant lines, not only will the genes of the two lines experience different mutations, but whole blocks of genes will be cut, pasted and sometimes inverted.
There is no reason a priori for a creator to adhere to a particular order for the genes, so one powerful way to create life in such a way as to be inconsistent with the prediction of common descent is to ensure that the synteny between related species was as low as possible. When geneticists examine the genetic data, what they find is however entirely consistent with common descent. In general, species that are believed to have a recent common ancestor have a higher synteny than those that have a remote common ancestor.
We can see this [6] in the fruit fly Drosophila, which have been extensively studied by geneticists for the best part of a century. When you examine similar portions of the genomes of Drosophila melanogaster and the closely related Drosophila ananassae, you can see that an entire block of genes has been inverted:

What's happened? There's been an inversion event, where a complete block of genes has been inverted:
 |
| Images based on data from Bhutkar, A., Schaeffer, S.W., Russo, S.M., Xu, M., Smith, T.F., and Gelbart, W.M. (2008). Chromosomal rearrangement inferred from comparisons of 12 Drosophila genomes. Genetics 179; 1657-1680 |
The geneticist Dennis Venema writes:
“Now imagine analyzing this for all twelve species and—in each case—examining all 14,000 (or so) genes. The position of every chromosome break in the time since the 12 species had a common ancestor has been mapped out. 40 million years of history has been all laid out showing the set of disruptions of the single file order in which the genes are stored. We even know about how often those disruptions occur in a lineage: breaks, like the two described above, take place about once every 200,000 years. This rate has been fairly constant in the approximately 40 million year history of these twelve lineages. Species that diverged only recently (judged by an independent mechanism) have only a small number of breaks and a large amount of synteny, On the other hand, species which diverged longer ago (again, as judged by an independent mechanism), have a much larger number of breaks and a smaller degree of synteny.” [7]
It is this strong consonance between evolutionary family trees generated from different lines of genetic evidence that makes the case for common descent so powerful. Conversely, the special creationist needs to justify why God created the genomic data of life in such a way that analysing syntenic data and gene homology produce evolutionary family trees that agree remarkably well with the standard family trees generated by morphology. None of this is essential for the function of each species. The possible number of ways in which to code for the same cytochrome c protein is unimaginably large, yet the genes cluster in a way predicted by common descent, while the gene order of related species could easily vary. If God has created life with genomic data, which when analysed shows a pattern consistent with common descent, it is quite reasonable to assume common descent is in fact true.
Shared genomic errors
Many people are quite surprised to discover that only a small part of the human genome is directly involved in coding for proteins or regulatory sequences. Most of it is non-coding DNA, consisting of broken genes, remnants of past retroviral infection and mobile genetic elements that do nothing other than paste themselves randomly into the genome. The breakdown of our genome [8] shows just how little is actually functional:
• Transposable elements
- 44% non-essential
- ~0.1% functional
• Viral elements
- 9% non-essential
- ~0.1% functional
• Pseudogenes
- 1.2% non-essential
- < 0.1% functional
• Ribosomal RNA genes
- 0.19% non-essential
- 0.22% essential
• Other RNA encoding genes
- ~ 2% essential
• Protein encoding genes
- 9.6% non-functional
- 1.8% essential
• Regulatory sequences
- 0.6% essential
• Origins of DNA replication
- < 0.1% essential
• Scaffold attachment regions
- < 0.1% essential
• Highly repetitive DNA
- 1% non-functional
- 2% essential
• Intergenic DNA
- 2% essential
- 26.3% non-conserved.
Nearly two thirds (65%) of our genome is non-functional junk, while less than one tenth (8.7%) is functional. The remaining ~ 26% has no known function, but based on what is known of its structure, it is most likely to be junk.
An often used analogy is to compare the genetic data in the nucleus of a cell to a large multi-volume encyclopaedia, but what modern genomics research is showing is that if our genome was represented by a 10-volume encyclopaedia, nearly seven of those would contain random gibberish and long stretches of repeated letters, while barely one volume would contain useful information. The presence of a large amount of non-functional ‘junk’ DNA is not proof of evolution, but it is a strong argument against design, particularly when one looks at the nature of that DNA. I’ll look at three of the most common class of non-functional DNA below.
Transposable elements, also known as transposons or more colloquially ‘jumping genes’ are DNA segments that are able to cut and paste themselves elsewhere in the genome. As this is done randomly, quite often the transposon can insert into a functional gene, rendering it non-functional and thereby causing harm to the organism. By chance, such an insertion event may result in benefit to the organism, but generally, the process is either deleterious or neutral.
Two classes of transposon exist; DNA transposons which I mentioned earlier, and RNA transposons or retrotransposons. These transposable elements are able to copy themselves to RNA via reverse transcription and then make a DNA copy which is randomly inserted elsewhere. As they are able to replicate themselves, retrotransposons can rapidly multiply copies of themselves and in the process increase the genome size. Most of the retrotransposons in the human genome are no longer able to copy and paste themselves. As with DNA transposons, retrotransposons can occasionally be co-opted by the genome, but as mentioned earlier, their action is either neutral or deleterious if they paste into a working gene and inactivate it. Transposons are parasitic elements which exist simply to propagate themselves, and are extremely difficult to reconcile with ‘intelligent design.’
Viral elements are remnants of previous viral infections that have integrated themselves into the host genome. The vast majority of thee viral elements originate from retroviruses which use RNA instead of DNA as their genetic material. When they infect a host cell, they make a DNA copy of themselves which is then able to be inserted into that cell’s DNA. If the virus infects the germline (that is, integrating into sperm or egg cell DNA) it will be able to be passed on to the next generation.
Pseudogenes are genetic elements that resemble genes but are not able to produce the product for which that gene normally would code. Three types of pseudogene exist: unitary, duplicated and processed.
Unitary: result when a normally functioning gene acquires a crippling mutation that prevents it from functioning. One of the better known unitary pseudogenes is the gulonolactone oxidase pseudogene, GULOP. Most organisms are able to make their own vitamin C, but in a number of species, most notably humans, apes, guinea pigs and bats, the gene which codes for L-gulonolactone oxidase, one of the enzymes involved in vitamin C biosynthesis is non-functional.
Duplicated: occurs when a gene is copied, and one or more of the copies becomes inactivated through mutation. The haemoglobin gene family is one notable example as there are a number of haemoglobin pseudogenes in addition to the functional haemoglobin genes, the former having arisen via duplication and inactivation.
Processed: these occur if the RNA transcript of a gene, which normally is used by the cell as the ‘recipe’ to create the protein for which it encodes, is copied via reverse transcriptase to a DNA copy and is inserted randomly into the genome. In order to appreciate how readily one can tell the difference between a processed pseudogene and the gene from which it originated, it is useful to know some of the details of genetics. A working gene will have a promoter element (a section of genetic material involved in initiating gene transcription), while the part of the gene that actually codes for the protein will be broken up with genetic elements called introns that are spliced out during transcription. The tail-end of the RNA copy of a gene will contain what is called a poly-A tail, which is important in stabilising the RNA copy. The processed pseudogene lacks introns and a promoter element, but has a poly-A tail added.
Common descent would predict that species descending from a common ancestor not only would have similar genes, but have most of those genes in the same order. Building on from the information above, we would also predict that if an organism was infected by a retrovirus that became fixed in its genome, had one of its genes incapacitated by mutation and converted to a pseudogene, or had a number of retrotransposition events occurring in its genome, then these ‘genetic errors’ would be inherited by species descended from the ancestral species.
Conversely, special creation would have no credible answer other than to claim that purely by chance:
- the same retrovirus inserted into the same location,
- the same gene became incapacitated by exactly the same mutation
- the same RNA transcript of a gene became reverse transcribed and pasted into the same location
- the same retrotransposon copied and inserted itself into the same location
The literature is replete with examples, but two should suffice. The first – and arguably most famous example – is that of the GULO-P pseudogene which in humans, chimpanzees, orangutans and macaques [9] is broken in exactly the same was. (For those interested in the details, the 164 nucleotide sequence of exon X shared a single nucleotide deletion). While guinea pigs also have a non-functional GULO gene, this was inactivated in a completely different way [10] and therefore represents a separate pseudogenisation event. The GULO pseudogene in humans, apes and old world monkeys is broken in exactly the same way, consistent with the incapacitation of GULO in the common ancestor of these primates.
After a gene is rendered non-functional, it becomes selectively neutral, which means that it is free to acquire random mutations. If we look at all the species that have the same pseudogene in common, those that share a recent common ancestor should differ by fewer random mutations. Conversely, those that have a distant common ancestor will have had more time to accumulate random mutations, and therefore will be less similar. By creating a family tree based on this mutation data, we should be able to create a tree roughly consistent with the expected evolutionary tree. This is exactly what we see when we look at the GULO data in primates:

The primate data clusters closely together, with humans and chimpanzees closest of all. Rodents cluster closely together, with guinea pigs clustering closely with the rodents. This is exactly what we’d expect if common descent was true. Special creation simply has no credible answer. Remember, when we look at the primate data, we’re looking at random mutations in a non-functional gene. The creationist either has to assume that this occurred purely by chance; the odds of which are so remote as to be practically impossible or answer:
- Why did God create primates with a broken vitamin C synthesis system, leaving others with the ability to synthesise vitamin C
- Why did God create humans, apes and old world monkeys (which according to evolutionary biology form a clade, or group of organisms sharing a common ancestor) with a GULO pseudogene broken in exactly the same way?
- Why did God then insert random mutations into this broken pseudogene in such a way as to allow one to construct an evolutionary family tree that agrees with the conventional evolutionary tree derived from morphological data?
“Both humans and chimps have a broken copy of a gene that in other mammals helps make vitamin C…It's hard to imagine how there could be stronger evidence for common ancestry of chimps and humans.” [11]
The second example is the large number of endogenous retroviruses (ERVs) shared by humans and other primates. ERVs are the remnants of previous retroviral infections that became fixed in the germ line and therefore were able to be inherited like other genetic elements. Almost all of them have been inactivated and degraded by deletions and mutations, which means they are no longer functional.
Common descent would predict that if a species was infected by a retrovirus which became fixed in the genome, all the descendant species should also inherit this ERV. The probability that multiple species have independently been infected by the same retrovirus at the same point in their genomes is quite remote. The respected virologist John Coffin notes:
“Because the site of integration in the genome, which comprises some three billion base pairs in humans, is essentially random, the presence of an ancient provirus at exactly the same position in different, but related, species cannot occur by chance, but must be a consequence of integration into the DNA of a common ancestor of all the species that contain it. It evolution of retroviruses follows, therefore, that we can infer what viruses were present millions of years ago by examining the distribution of endogenous proviruses in modern species.” [12]
ERVs are in addition very tightly integrated into the genome, making it extremely hard for them to be neatly excised without leaving behind remnants:
Therefore, an ERV locus shared by two or more species is descended from a single integration event and is proof that the species share a common ancestor into whose germ line the original integration took place. Furthermore, integrated proviruses are extremely stable: there is no mechanism for removing proviruses precisely from the genome, without leaving behind a solo LTR or deleting chromosomal DNA. The distribution of an ERV among related species also reflects the age of the provirus: older loci are found among widely divergent species, whereas younger proviruses are limited to more closely related species. [13]
It’s hard to get any more definite than that. The chances of the same retrovirus inserting itself into the same location in the genomes of a human and a chimpanzee is around 1 in 3,000,000,000. If we have multiple ERVs found in the same places in related species, then the odds of this occurring by chance become so remote as to be impossible.
This is exactly what we see when we examine human and primate genomes - multiple ERVs inserting at the same place in their respective genomes. More importantly, the pattern of insertion of these ERVs matches the standard evolutionary family tree. Medstrand and Mager [14] examined the pattern of insertions of a particular class of endogenous retroviruses, the HERV-K family. Thirty-seven ERV fragments were aligned into clusters based on sequence divergence. When they compared this with primate genomic data, they found that the clusters with greater divergence were also found in Old World monkeys and apes, while those with a lesser amount of divergence were found only among gorillas and chimpanzees. The cluster with the least amount of divergence was found only in the human genome.
 |
| Approximate integration times of HERV-K elements. Arrows indicate the lineage in which a particular LTR was first detected, and numbers refer to the HERV clusters |
The pattern of HERV-K element insertion shown above matches what common descent would predict.
In general, LTR sequences of clusters 1 to 5 were first identified in Old World monkey and gibbon DNAs, whereas LTRs of cluster 8 first appeared in DNAs of gorilla and chimpanzee. For example, the AF001550 LTR of cluster 3 is not present in Old World monkeys but is present in gibbon and all higher primates. In contrast, the AC003023 cluster 8 LTR is found only in chimpanzee and human, indicating a more recent integration, Initial results with primers flanking three of the integrated LTRs of cluster 9 resulted in the expected amplification products in human DNA but not in any of the other primate DNAs, To demonstrate that sequences of cluster 9 were unique to human DNA, primers flanking the other six identified LTRs of this cluster, including the full-length HERV-K10 element, were used in the amplification of primate DNA. Indeed, all were detected only in human DNA, indicating that sequences derived from this cluster integrated after the divergence of the human lineage from the great apes. [15]
It is this nesting of HERV clusters, in a way according with what common descent predicts that makes this powerful evidence for common descent. ERVs are evidence of ancient retroviral infection, and the presence of the same ERV at the same place in related species is as Coffin states prima facie evidence for an ancient retroviral infection in the ancestor of both species. When we multiply the number of ERVs that have integrated in the same place in many primate genomes, but do so in the pattern above, the case for common descent based solely on ERV inclusions becomes overwhelming.
Synteny and the Origin of Chromosome 2
The genomes of humans and the great apes are highly syntenic, which is what common descent would predict if they shared a recent common ancestor. Even at the level of chromosomal banding, the marked similarity between apes and humans is obvious:
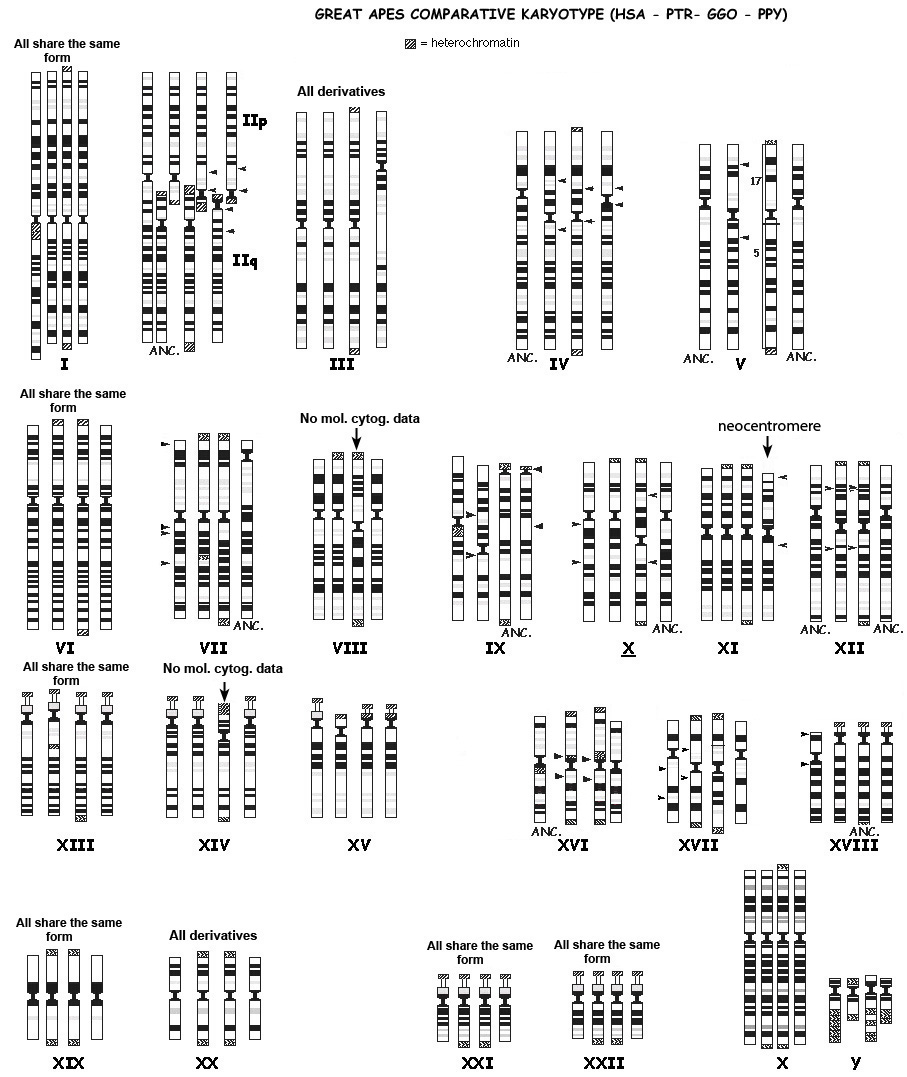 |
| Source: http://www.biologia.uniba.it/primates/1-GA/2-Comparative-Yunis.html |
However, humans have only 23 pairs of chromosomes, while the great apes have 24. If common descent was true, we’d expect to see evidence that human chromosome 2 was the result of a fusion of two chromosomes homologous to ape chromosomes. As one can see above, the banding pattern of chromosome 2 is strongly similar to those of two ape chromosomes. As long ago as 1982, Yunis and Prakash wrote:
Evidence for a common ancestor of man and chimpanzee also comes from chromosome 2, since human chromosome 2 is most simply explained by telomeric fusion of a chimpanzee-like 2p chromosome and a 2q chromosome similar to that of chimpanzee and gorilla. [16]
Since the early 1980s, the discipline of molecular biology has exploded to the point where genome sequencing is becoming common place. We are now in a position to look for evidence of such chromosomal fusion at the molecular level. Specifically, we would expect to see evidence of both telomeric and centromeric remnants in the body of human chromosome 2.

Twenty years ago, Idjo et al analysed chromosome 2, and found proof of a telomere to telomere fusion event:
We have isolated two allelic genomic cosmids that were localized to chromosome 2, each containing two arrays of telomeric repeat TTAGGG in an inverted arrangement. Flanking sequences are characteristic for the preterminal regions of human chromosomes. The data we present here demonstrate that a telomere-to-telomere fusion of ancestral chromosomes occurred, leaving a pathognomonic relic at band 2q13. This fusion accounts for the reduction of 24 pairs of chromosomes in the great apes (chimpanzee, orangutan, and gorilla) to 23 in modern human and must, therefore, have been a relatively recent event. Comparative cytogenetic studies in mammalian species indicate that Robertsonian changes have played a major role in karyotype evolution. This study demonstrates that telomere-telomere fusion, rather than translocation after chromosome breakage, is responsible for the evolution. [17]
In addition, centromeric remnants have been identified in human chromosome 2, which again is consistent with what we would expect if common descent was true:
In this paper we demonstrate centromeric repeats at 2q21 that could be the remains of the ancestral centromere. This is supported by the fact that pαH21 hybridizes at the centromeres of both chromosomes that are the origin of human chromosome 2. In Fig. 1F a dual labeling hybridization with clones pαH21 and c8.1 (identifying the fusion point, Ijdo et al. 1991) shows the relative location of the remnants of the two chromosomal structures. [18]
Space precludes further elaboration of examples of how comparative genomics provide overwhelming support for common descent; the literature on this subject [19-21] is comprehensive, and leaves the objective reader in little doubt as to the fact of common descent.
References
1. Watts J "Evolution or Creation? The Argument From Comparative Anatomy" The Testimony January 2005 p 31-33
2. Technically, we refer to orthologous genes if they arose via a speciation event and paralogous genes if they arose via gene duplication.
3. There are some minor variations in the genetic code, but these do not affect the argument and are not dealt with here. See any biochemistry textbook for the details.
4. A nucleotide consists of one of the four nucleobases (adenine, cytosine, guanine and uracil), a single phosphate group and a sugar. In DNA, uracil is replaced by thymine.
5.Max E.E. "Plagiarized Errors and Molecular Genetics"
6. Venema D “Signature in the Synteny” Science and the Sacred April 19th 2010 The following discussion on synteny is heavily indebted to his approach.
7. loc cit.
8. Moran L “What’s in Your Genome?” Sandwalk May 8th 2011
9. Ohta Y, Nishikimi M "Random nucleotide substitutions in primate nonfunctional gene for L-gulono-gamma-lactone oxidase, the missing enzyme in L-ascorbic acid biosynthesis.” Biochim Biophys Acta. 1999 Oct 18;1472(1-2):408-11.
10. Nishikimi M, Kawai T, Yagi K. "Guinea pigs possess a highly mutated gene for L-gulono-gamma-lactone oxidase, the key enzyme for L-ascorbic acid biosynthesis missing in this species." J Biol Chem. 1992 Oct 25;267(30):21967-72.
11. Behe M “The Edge of Evolution. The Search for the Limits of Darwinism” (2007, Free Press) pp 71-72
12. Coffin JM “Evolution of Retroviruses: Fossils in our DNA” Proceedings of the American Philosophical Society (2004) 148:3, 264-280
13. Johnson WE Coffin JM Constructing primate phylogenies from ancient retrovirus sequences Proc. Natl. Acad. Sci. USA (1999) 96:10254-10260
14. Medstrand P, Mager DL “Human-Specific Integrations of the HERV-K Endogenous Retrovirus Family” J Virol (1998) 72(12):9781-9787
15. ibid, p 9784
16. Yunis, J.J. and Prakash, O. ‘The origin of man: a chromosomal pictorial legacy’ Science (1982) 215, 1525
17. Ijdo, J.W., Baldini, A., Ward, D.C. et al. ‘Origin of human chromosome 2: an ancestral telomere-telomere fusion’, Proc Natl Acad Sci USA (1991) 88, 9051.
18. Baldini, A., Ried, T., Shridhar, V. et al. ‘An alphoid DNA sequence conserved in all human and great ape chromosomes: evidence for ancient centromeric sequences at human chromosomal regions 2q21 and 9q13’, Hum Genet (1993) 90, 577.
19. Koonin EV “The Logic of Chance: The Nature and Origin of Biological Evolution” (2011 FT Press)
20. Lynch M “The Origins of Genome Architecture” (2007 Sinauer Associates)
21. Gregory TR (Ed) “The Evolution of the Genome” (2005 Elsevir Academic Press)
3. There are some minor variations in the genetic code, but these do not affect the argument and are not dealt with here. See any biochemistry textbook for the details.
4. A nucleotide consists of one of the four nucleobases (adenine, cytosine, guanine and uracil), a single phosphate group and a sugar. In DNA, uracil is replaced by thymine.
5.Max E.E. "Plagiarized Errors and Molecular Genetics"
6. Venema D “Signature in the Synteny” Science and the Sacred April 19th 2010 The following discussion on synteny is heavily indebted to his approach.
7. loc cit.
8. Moran L “What’s in Your Genome?” Sandwalk May 8th 2011
9. Ohta Y, Nishikimi M "Random nucleotide substitutions in primate nonfunctional gene for L-gulono-gamma-lactone oxidase, the missing enzyme in L-ascorbic acid biosynthesis.” Biochim Biophys Acta. 1999 Oct 18;1472(1-2):408-11.
10. Nishikimi M, Kawai T, Yagi K. "Guinea pigs possess a highly mutated gene for L-gulono-gamma-lactone oxidase, the key enzyme for L-ascorbic acid biosynthesis missing in this species." J Biol Chem. 1992 Oct 25;267(30):21967-72.
11. Behe M “The Edge of Evolution. The Search for the Limits of Darwinism” (2007, Free Press) pp 71-72
12. Coffin JM “Evolution of Retroviruses: Fossils in our DNA” Proceedings of the American Philosophical Society (2004) 148:3, 264-280
13. Johnson WE Coffin JM Constructing primate phylogenies from ancient retrovirus sequences Proc. Natl. Acad. Sci. USA (1999) 96:10254-10260
14. Medstrand P, Mager DL “Human-Specific Integrations of the HERV-K Endogenous Retrovirus Family” J Virol (1998) 72(12):9781-9787
15. ibid, p 9784
16. Yunis, J.J. and Prakash, O. ‘The origin of man: a chromosomal pictorial legacy’ Science (1982) 215, 1525
17. Ijdo, J.W., Baldini, A., Ward, D.C. et al. ‘Origin of human chromosome 2: an ancestral telomere-telomere fusion’, Proc Natl Acad Sci USA (1991) 88, 9051.
18. Baldini, A., Ried, T., Shridhar, V. et al. ‘An alphoid DNA sequence conserved in all human and great ape chromosomes: evidence for ancient centromeric sequences at human chromosomal regions 2q21 and 9q13’, Hum Genet (1993) 90, 577.
19. Koonin EV “The Logic of Chance: The Nature and Origin of Biological Evolution” (2011 FT Press)
20. Lynch M “The Origins of Genome Architecture” (2007 Sinauer Associates)
21. Gregory TR (Ed) “The Evolution of the Genome” (2005 Elsevir Academic Press)


Comments
Post a Comment